Freezing and thawing procedures for successful cryopreservation
The physical and biological effects of a cooling process can be tolerated, without injury, by a limited range of specimens but the great majority would suffer lethal injury without some form of protective protocol being employed. Protection against such injury is best achieved by the combined use of controlled ice nucleation, chemical protectants (cryoprotectants) as additives and the appropriate manipulation of cooling and warming rates.
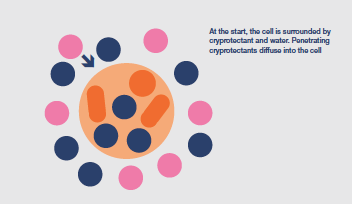
Cryoprotectant chemicals are added to the cells immediately before beginning the preservation protocol. They penetrate the cells, to a greater or lesser extent, inhibit intracellular ice formation and protect against the damaging effects of an increasingly concentrated cytoplasm.
Cryoprotectants may be toxic, in varying degrees, to the cells and timing of their use, concentration and application temperature have to be balanced against survival benefit.
Common cryoprotectants include dimethyl sulfoxide, ethanediol, glycerol, and propanediol.

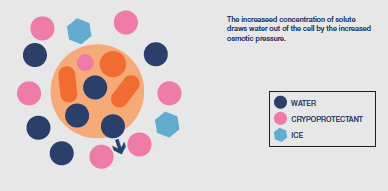
Immediately ice is nucleated in the extracellular matrix the cells undergo a degree of potentially damaging osmotic shock as the solutes have become concentrated into the residual liquid
surrounding the cells. This enhanced, external concentration of solutes causes significant movement of water out of the cells leading to dehydration injury (solution effects). However, the risk of injury can be minimised by ensuring that ice is nucleated at the earliest possible temperature, producing the lowest possible level of initial osmotic stress.
This induced nucleation is necessary as solutions readily become super-cooled, falling below their nominal freezing point but without the formation of ice e.g. water will undergo the phase change to ice if nucleated just a little below 0°C, yet can remain unfrozen (super cooled) until -40°C below zero. If significant super-cooling occurs before nucleation a high, instant level of potentially lethal osmotic stress can be expected. There are various mechanical ways of inducing nucleation including rapid agitation of cooled vials, touching a deep-cooled
implement to the outside of the freezing container (e.g. forceps with tips cooled in LN) or, briefly accelerating the cooling rate (a cooling ‘dip’). Some programmable freezers have an
attachment to effect nucleation. Also commercially available are various non-toxic additives that will induce nucleation at the highest possible temperature in suitably cooled solutions.
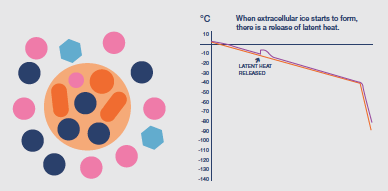
Once ice is nucleated the temperature of the sample will, inevitably, begin to rise due to the latent heat liberated during ice formation (an exotherm). If uncontrolled, this temperature
rise can be damaging, as the proportion of ice in the system fluctuates; so the cooling protocol should be adapted to minimize this rise, typically by an increase in cooling rate at least until the latent heat exotherm is eliminated.
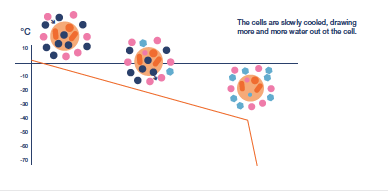
After the extracellular environment is nucleated the next challenge in the protocol is to control the rate of cooling to ensure that the necessary amount of protective cellular dehydration occurs. There will be slow cooling rates that produce too much osmotic stress due to the length of exposure to extracellular ice and rapid cooling rates that limit dehydration and raise the probability of lethal intracellular ice formation to unacceptable levels. In between will be an ’optimal’ cooling rate that holds both these stresses at acceptable levels.
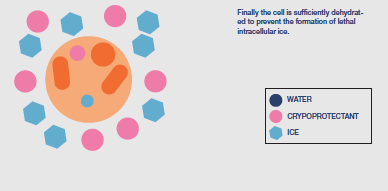
Cooling rate may need to be varied during the protocol, which will have to be determined for each different cell type. The objective is to remove sufficient water from the cell to allow
water in the concentrated cytoplasm to vitrify as cooling progresses i.e. form a less injurious ‘glass’ rather than the highly damaging, crystalline ice. Typically, cooling to achieve
dehydration is continued to a base temperature between -35 and -45°C before the sample is immersed directly into LN.
In most situations rapid warming is essential for good cell survival, with sub-optimal rates increasing the probability of intracellular ice formation as the sample thaws, with inevitable
loss of viability. Smaller sample containers, such as cryovials, can be immersed in water at 37°C and visually monitored until the last ice crystals have melted. Small variations from the
required warming profi le can cause signifi cant loss of viability and so samples must be transferred directly from LN into the warming bath. The volume and heating control of the bath
must avoid any signifi cant reduction in temperature due to the introduction of the sample. Once thawed a sample may need to be rinsed to remove cryoprotectants and may also need time to repair injuries. A reduced incubation temperature, avoidance of shaking and storage in the dark may all be helpful at this stage.
A number of semi-controlled devices to assist and monitor the thawing process are coming onto the market.
Viability should be assessed immediately post-thaw and monitored frequently thereafter as the level may decline as damaged cells may fail to repair and recover. In particular, enzyme-linked histochemical assays can overestimate survival as active enzymes can persist in damaged cells that cannot repair and, subsequently, die. Documentation for the frozen sample should indicate the expected performance on recovery e.g. levels of enzyme activity, and it is only when this has been achieved that final ‘survival’ can be calculated.